Sample Progression Discovery (SPD) version 2
Peng Qiu1, Andrew Gentles2,
Sylvia Plevritis2
1Department of Bioinformatics and Computational Biology, University
of Texas MD Anderson Cancer Center
2Department of Radiology, Stanford University
We present a novel computational approach, Sample
Progression Discovery (SPD), to discover patterns of biological progression
underlying a microarray dataset. In contrast to the majority of microarray data
analysis methods which focus on identifying differences between sample groups
(i.e. normal vs. cancer, treated vs. control), SPD aims to identify an
underlying progression among individual samples, both within and across sample
groups. We applied SPD to gene expression data of cell cycle, B-cell
differentiation, and mouse embryonic stem cell differentiation, where the
underlying progression was known but hidden from SPD. We show that SPD correctly
identified the progression among samples and the gene modules that are
associated with the progression. We view SPD as a hypothesis generation tool,
when applied to datasets where the progression is unclear. For example, when
applied to a microarray dataset of cancer samples, SPD would assume that the
cancer samples collected from individual patients represent different stages
during an intrinsic progression underlying cancer development. The inferred
relationship among the samples may therefore indicate a pathway or hierarchy of
cancer progression, which serves as a hypothesis to be tested.
The algorithm is implemented in Matlab 7, with a Graphic User Interface on top
of it, designed and written by Peng Qiu.
A manuscript of this work is available: Peng Qiu, Anderw Gentles and Sylvia Plevritis, "Discovering Biological Progression underlying Microarray Samples", PLoS Computational Biology, vol. 7, issue 4, e1001123, 2011.
An older versino of the software is available at here - SPD_v1
If you have any questions or find any problems in it, please email me at peng.qiu@bme.gatech.edu
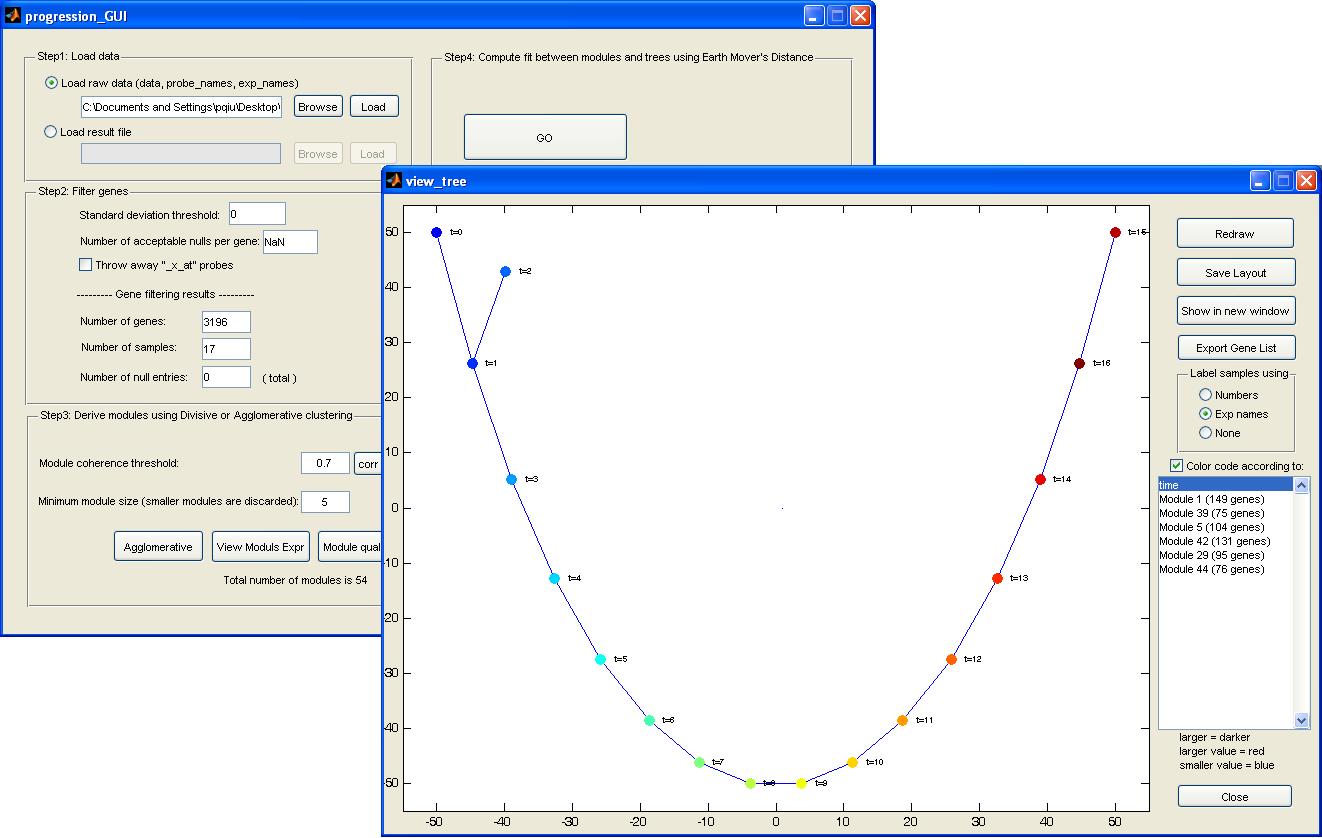
Here is a screen shot of our software
Installation instructions:
This package requires Matlab 7. In order to give users
maximum freedom of manipulating this software, the raw .m files are provided.
(1) download the zip package: SPD.zip
(last updated on Dec 01, 2013)
(2) unzip to your local machine
(3) open Matlab 7 and change the directory to where the package is unzipped, or add that directory to matlab path
(4) type "progression_GUI" and enter,
the GUI will show up
We also provide one example data and result file: SPD_example_data_files.zip
License conditions:
The SPD software is free for academic use. A patent for SPD has been applied for on behalf of Stanford University. For license conditions, please contact the Office of Technology Licensing at Stanford (Kirsten Leute, kirsten.leute@stanford.edu).
User Manual:
1. Prepare input file:
Input file contains at least 3 variables:
probe_names : n * 1 cell array, each element is the name of one gene/feature
exp_names : 1 * m cell array, each element is the name of one sample/array
data : n * m expression data matrix
Two optional variables can be used to input clinical information or other sample
annotations
color_code_names: k * 1 cell array
color_code_vectors: k * m matrix that stores clinical info
For example:
color_code_names = [ {'Gender'};
{'Tumor Grade'};
];
color_code_ vectors= [ 0 0 0 0 0 0 1 1 1 1 1 0 0 1 1;
1 2 3 2 2 2 1 1 3 3 3 2 1 1 1;
];
An input file can contain other variables, but the software will just ignore
them.
See examples in: SPD_example_data_files.zip
2. Load data
Users can load (1) a raw input data file defined above, or (2) a result file
generated by the software.
3. Filter genes
Filter genes according to user-defined threshold of standard deviation, and
number of acceptable nulls per gene. Any genes with smaller STD or more NULL
entries are excluded. A user can also choose to exclude "_x_at" probesets.
The software shows some basic information after the filtering: number of genes
left, number of samples, number of mull entries.
After filtering genes, null entries are imputed / filled using KNN imputation,
so that the subsequent steps do not have to worry about null entries.
If more sophisticated filters are needed, users can apply those filters when
preparing the input file, and then ignore the gene filtering functions in the
GUI.
4. Clustering genes (Step 1 in SPD)
Button "Agglomerative" starts a button-up agglomerative clustering algorithm.
The stopping criterion of the clustering method is user-defined, which is the
desired module coherence. The default is 0.7, but this could be
dataset-dependent. The button "corr hist" can be used to view a histogram of all
the pair-wise correlation (after genes are filtered). If the histogram has a
heavy tail, meaning many gene pairs share high correlation, higher coherence
parameter might be more appropriate.
The parameter "Minimum module size" can be used to exclude small clusters.
After the clustering algorithm finishes, the two buttons
"view modules Expr" and "Model quality" are enabled. You can use these two
buttons to visualize the clustering results.
The previous verion has another clustering algorithm, but that one was removed in this version.
5. Construct MSTs and compare modules and MSTs (step 2 and step 3 in SPD)
The "GO" button does the following:
(1) Construction one MST based on each gene module
(2) Compute the earth mover's distance between all the modules
and all the trees.
6. Identify modules similar in terms of progression (step 4 in SPD)
To generate a progression-similarity matrix between gene modules, the user needs to define a threshold that determines whether the fit between a module and a tree is significant. The default value 0.05 means that, among all the module-tree pairs, the top 5% with most significant earth mover's distances are considered to "fit well with each other", and are used to construct the progression similarity matrix PSM (details about how to construct the PSM is available in our PLoS CB paper).
Then, click the button "Show gene module adjacency", a figure will pop up, which
shows the progression similarity matrix, similarity between modules in terms of progression.
We suggest the user to vary the threshold among 0.05, 0.1, 0.15, and see whether a significant block can be observed.
Visually identify (high value) blocks along the diagonal. It is possible that
there exist more than one block along the diagonal line.
Zoom-in this pop up window to see which modules are associated to the visually
identified block(s).
Manually input the identified modules (a space-separated string that contains the IDs of the modules in the identified block) and click "add". After clicking "add", one
entry will be shown in the list-box. This entry is one progression pattern
defined by the modules that the user just "add"ed. (Multiple progression
patterns can be added, each correspond to one block that the user visually
identified).
To view the progression pattern, click "view progression". Another window will
pop up. If the number of samples is smaller than 50, this step will take ~ 30 sec.
If the number of samples is large, this step may take long, because my tree-visualization algorithm is slow.
In the pop-up window:
(1) Nodes/samples can be dragged around.
(2) After moving the nodes, the new layout can be saved. Then next time when the
user views this progression pattern, the saved layout will be displayed, instead
of running my slow tree-visualization algorithm. To save this new layout, you
need to click the "Save Layout" button in the view_tree window, and the "Save
Results" button in the main window.
(3) Nodes can be color-coded according to the modules that support this
progression pattern (blue means low value, red means high value).
(4) Nodes can also be color-coded by the clinical information from the input
data file: color_code_names, and color_code_vectors.
(5) Button "export gene list" writes two files in the current directory. Both
contain the same information, the list of genes in the modules that support the
progression.
NOTE1: the major differences between this version and the previous version of the software are: (1) how the module-tree fit is computed, and (2) the threshold used to construct the progression similiarity matrix.
NOTE2: this version requires one matlab function (linprog) which should be in the optimization toolbox of matlab. If your matlab version does not have this function/toolbox, you will see an error saying that linprog function is undefined.